Синдром Хантера: коротко про головне

Синдром Хантера: коротко про головне
Синдром Хантера — це рідкісне генетичне захворювання1, що належить до лізосомних хвороб накопичення. Генетична мутація призводить до порушення синтезу ферменту лізосом і, як наслідок, порушення розщеплення особливих молекул – ГАГ*2.
*ГАГ — глікозаміноглікани.

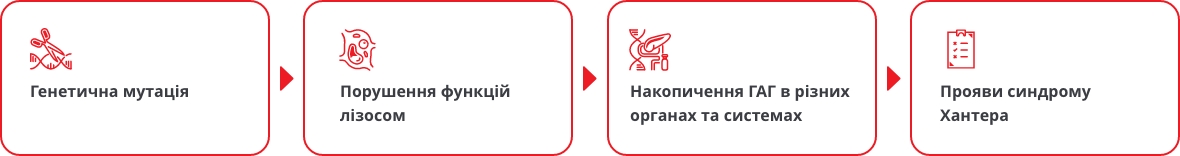
Діти з с. Хантера народжуються здоровими на вигляд, але накопичення ГАГ призводить до появи перших проявів с. Хантера — зазвичай у віці 2-4 років2. Ранні ознаки синдрому Хантера часто неспецифічні і можуть нагадувати звичайні скарги, не викликаючи підозри у лікарів3.

Педіатри3 та отоларингологи4 — перші спеціалісти, до яких може потрапити дитина з ранніми проявами с. Хантера
Знання лікарем основних проявів с. Хантера може врятувати життя цих пацієнтів!
На сьогодні в Україні доступне лікування с. Хантера з доведеною клінічною ефективністю5,6. Своєчасна діагностика та ранній початок лікування продовжують тривалість життя!7
- Francesca D'Avanzo et al. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. Int J Mol Sci . 2020 Feb 13;21(4):1258.
- Burton BK, Giugliani R. Diagnosing Hunter syndrome in pediatric practice: practical considerations and common pitfalls. Eur J Pediatr 2012; 171(4): 631–639.
- Scarpa M et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis 2011; 6(1): 72.
- Muenzer J et al. Multidisciplinary Management of Hunter Syndrome. PEDIATRICS 2009; 124(6): e1228–e1239.
- Muenzer J., Wraith J.E. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genetics in Medicine. August 2006 Vol. 8 No. 8.
- Giugliani R., Hwu W.-L. Multicenter, open-label study evaluating safety and clinical outcomes in children (1.4–7.5 years) with Hunter syndrome receiving idursulfase enzyme replacement therapy. Genetics in medicine, Volume 16, Number 6, June 2014.
- Burton B.K, Lego V. Survival in idursulfase-treated and untreated patients with mucopolysaccharidosis type II: data from the Hunter Outcome Survey (HOS). Inherit Metab Dis. 2017; 40(6):867-87.